
W patogenezie rozwoju cukrzycy typu 2 insulinooporność i niewydolność komórek beta trzustki odgrywają kluczową rolę. W wielu dotychczas przeprowadzonych badaniach wykazano również pewne powiązania pomiędzy nieprawidłową gospodarką żelazową a cukrzycą.
Cukrzyca to plaga XXI wieku. Schorzenie to charakteryzuje się podwyższonym poziomem glukozy we krwi. Niestety u osób z cukrzycą wzrasta także ryzyko rozwoju powikłań sercowych, udaru mózgu, niewydolności nerek, ślepoty, czy też rozwoju zespołu stopy cukrzycowej. W patogenezie rozwoju cukrzycy typu 2 insulinooporność i niewydolność komórek beta trzustki odgrywają kluczową rolę. W wielu dotychczas przeprowadzonych badaniach wykazano również pewne powiązania pomiędzy nieprawidłową gospodarką żelazową a cukrzycą. Dowiedziono mianowicie, iż przeładowanie żelazem jest ważnym elementem w patogenezie rozwoju cukrzycy[1].
Gospodarka żelazowa odgrywa istotną rolę zarówno w stanie zdrowia chorego, jak również w zakresie samej choroby. Jak wiadomo, żelazo bierze udział w wiązaniu i transportowaniu tlenu z płuc do tkanek obwodowych, w usuwaniu dwutlenku węgla, w procesie regulacji wzrostu i różnicowania komórkowego, a także w procesie transportu elektronów w mitochondriach. Uczestniczy także w syntezie DNA oraz w wielu różnych procesach metabolicznych.
Informacje na temat związku między zaburzoną gospodarką żelazową (przeładowaniem żelazem) a ryzykiem rozwoju cukrzycy pojawiły się już w XIX wieku. Dotyczyły one chorych z hemochromatozą[2,3]. Zwykle cukrzyca u tych osób była określana mianem ?diabetes bronze? ze względu na kolor zabarwienia skóry. Chorobowość z powodu cukrzycy u osób z hemochromatozą waha się od 22 do 63 proc.[4,5]. Również u chorych z talasemią, u których przeprowadzono liczne transfuzje krwi wykazano podobną zależność[6]. Chorobowość z powodu cukrzycy u chorych z talasemią po przetoczeniu krwi waha się od 6-14 proc. Wykazano także, iż podaż leków helatujących żelazo zapobiega rozwojowi zmian w trzustce, rozwojowi nietolerancji glukozy i cukrzycy[7].
Pragnę w tym miejscu dodać, że suplementacja żelaza u kobiet w ciąży nie zwiększa ryzyka rozwoju cukrzycy ciążowej. Również normalizacja hemoglobiny (po podaży żelaza) u chorych na cukrzycę typu 2 z przewlekłą chorobą nerek jest bezpieczna.
W badaniach typu case-control przeprowadzonych do chwili obecnej wykazano znamienną korelację pomiędzy zwiększonymi zapasami żelaza w organizmie a ryzykiem rozwoju cukrzycy. Nie wykazano natomiast takiej zależności między ryzykiem rozwoju cukrzycy a poziomem żelaza we krwi[8]. Zwiększone zapasy żelaza w organizmie przez wielu naukowców określane są w różny sposób. Najlepiej zastosować w tym celu określenie stężenia sTfR. Znajdująca się w krążeniu forma receptora tranferyny sTfR jest wynikiem eksternalizacji sTfR z komórek podczas cyklu endocytozy. Stężenia sTfR jest markerem erytropoezy. Wysokiemu stężeniu ferrytyny towarzyszy obniżenie poziomu sTfR.
Wydaje się, że najlepszym sposobem jest określenie stosunku rozpuszczalnego fragmentu receptora trasferyny do stężenia ferrytyny (stfR/ferrytyna). U osób przeładowanych żelazem wartość tego stosunku jest niższa niż u osób zdrowych[1]. Wykazano również, że z ryzykiem rozwoju cukrzycy koreluje poziom ferrytyny w surowicy krwi, sTfR, stosunek sTfR/ferrytyna oraz wysycenia transferyny.
Polimorfizmy genów biorących udział w regulacji gospodarki żelazowej a predyspozycja do rozwoju cukrzycy
Wiele genów biorących udział w homeostazie gospodarki żelazowej w organizmie odgrywa również rolę w gospodarce energetycznej ustroju. Dowiedziono, że polimorfizmy poniżej wymienionych genów wiążą się ze zmianą ryzyka rozwoju cukrzycy. Najważniejsze spośród polimorfizmów genów gospodarki żelazowej wpływających na ryzyko rozwoju cukrzycy prezentuje tabela.[9,10,11]
Tabela. Polimorfizmy genów gospodarki żelazowej wpływające na ryzyko rozwoju cukrzycy
| Lp. | Gen | Białko syntetyzowane | SNP | Ryzyko |
| 1. | TFRC | Receptor transferyny | rs3817672 (S1426) | ? |
| 2. | HFE | Białko hemochromatozy | rs1800562 (C2824) rs1799945 (H63D) | ? |
| 3. | HMOXI | Oksygeneza hemu | rs3074372 (Long GTu) | ? |
| 4. | TMPR556 | Osoczowa matriptaza | rs855791 (A736V) rs4820268 (D521) | ? |
| 5. | SMAD7 | Bloker SMAD | rs3764482 (S282F) | ? |
Przeprowadzone badania genetyczne jednoznacznie dowiodły, że w patogenezie rozwoju cukrzycy u osób z przeładowaniem żelazem należy brać pod uwagę obecne zmiany w budowie kilku genów. Najważniejsze z tych zmian prezentuję w tabeli. Przedstawiony zestaw zmian genetycznych nie wyczerpuje tego zagadnienia, a jedynie wskazuje na istotną rolę zmian genetycznych dotyczących zaburzeń w gospodarce żelazowej w patogenezie rozwoju cukrzycy.
Znaczenie żelaza w regulacji czynności komórek beta trzustki
Żelazo ma duże znaczenie w regulacji czynności komórek beta trzustki. Na czym ono polega? Żelazo odgrywa kluczową rolę w transporcie elektronów w mitochondriach, co jest niezbędne dla prawidłowej syntezy ATP. Proces ten jest czynnikiem cynglowym w pobudzaniu przez glukozę wydzielania insuliny (GSIS). Ponadto, żelazo jest czynnikiem pobudzającym syntezę wolnych rodników tlenowych, a tym samym czynnikiem nasilającym stres oksydacyjny. Wolne rodniki tlenowe zapoczątkowują i regulują pobudzaną przez glukozę syntezę insuliny. Podwyższony poziom glukozy zwiększa natomiast stężenie hepcydyny i obniża poziom żelaza. Tak więc, żelazo odgrywa bardzo istotną rolę w regulacji gospodarki węglowodanowej.
Jaki jest wpływ przeładowania żelazem na czynność komórek beta trzustki?
Komórki beta trzustki są bardzo wrażliwe na zaburzenia w homeostazie żelaza. Jest wiele czynników mających na to wpływ. Jakie to czynniki?
Po pierwsze, ekspresja enzymów działających antyoksydacyjnie (dysmutaza ponadtlenowa, peroksydaza glutationu i inne) jest bardzo niska w trzustce. Z tego powodu komórki te są bardziej wrażliwe na wzrost stężenia żelaza i generowany przez niego stres oksydacyjny. Komórki beta trzustki mają bardzo małą ekspresję ferroportyny oraz dużą ekspresję DMT1. Czyni to komórki beta bardziej wrażliwymi na podwyższony poziom żelaza. Poziom ferrytyny ujemnie skorelowany jest z czynnością komórek beta[12].
Indukowany niedotlenieniem czynnik 1? (HIF-1?) w komórce beta bierze udział w regulacji gospodarki węglowodanowej. Zależna od podwyższonego stężenia żelaza hydroksylacja oraz degradacja indukowanego niedotlenieniem czynnika (HIF-1?) prowadzi do zahamowania, stymulowanej glukozą, syntezy ATP. Wykazano, że w umiarkowany sposób podwyższony poziom HIF-1? istotnie poprawia wydzielanie insuliny i całą gospodarkę węglowodanową[13]. Hipoksja w komórkach beta prowadzi do spadku w cytoplazmie Zn2+ oraz zmniejsza ekspresję SLC30A8. Wzrost poziomu HIF-1? przywraca poziom Zn2+ do normy. Wzrost poziomu Zn2+ jest niezbędny dla syntezy insuliny. Nadmiar żelaza hamuje syntezę HIF-1? i poprzez to hamuje syntezę insuliny.
Jaki jest wpływ przeładowania żelazem na insulinooporność?
Wątroba jest głównym rezerwuarem żelaza w organizmie. Żelazo do hepatocytów dostaje się z krwi po związaniu z receptorem TfR1. Hepatocyty regulują z kolei gospodarkę żelazem w ustroju przy udziale białka hepcydyny. Hepcydyna pobudza fosforylację, interalizację oraz degradację ferroportyn. Czynnik ten hamuje wątrobową absorpcję żelaza oraz uwalnianie żelaza z makrofagów.
Wpływ przeładowania żelazem na insulinooporność nie został w pełni poznany. Tym niemniej przeprowadzono bardzo wiele badań, które przynajmniej częsciowo dają odpowiedź na postawione powyżej pytanie.
Jak wynika z tych badań, przeładowanie żelazem powoduje:
1. zwiększenie aktywności prozapalnej makropagów. Po pobudzeniu przez żelazo syntetyzują one w nadmiarze m.in. TNF-?, IL-6 i IL-1?[14]. Podwyższony poziom tych cytokin hamuje uwalnianie adiponektyny przez adipocyty. Adiponektyna w bardzo istotny sposób poprawia insulinowrażliwosć. Spadek jej stężenia powoduje narastanie insulinooporności[14];
2. zmniejszenie syntezy osteokalcyny przez komórki kostne. Białko to jest silnym czynnikiem pobudzającym uwalnianie adiponektyny. Zmniejszenie stężenia tego białka powoduje jednoczesne zmniejszenie stężenia adiponektyny i poprzez to narastanie insulinooporności[15];
3. zahamowanie fosfozylacji receptorów insulinowych w tkankach obwodowych takich jak hepatocyty, adipocyty czy miocyty. Wszystko, co powoduje nasilenie insulinooporności[1];
4. Produkowane przez zmienione prozapalnie cytokiny (TNF-?, IL-6 i IL-1?) wpływają również bezpośrednio hamująco na fosforylację receptorów insulinowych w tkankach obwodowych, a tym samym nasilają insulinooporność[1].
Podsumowanie powyższych procesów prezentuje rycina.
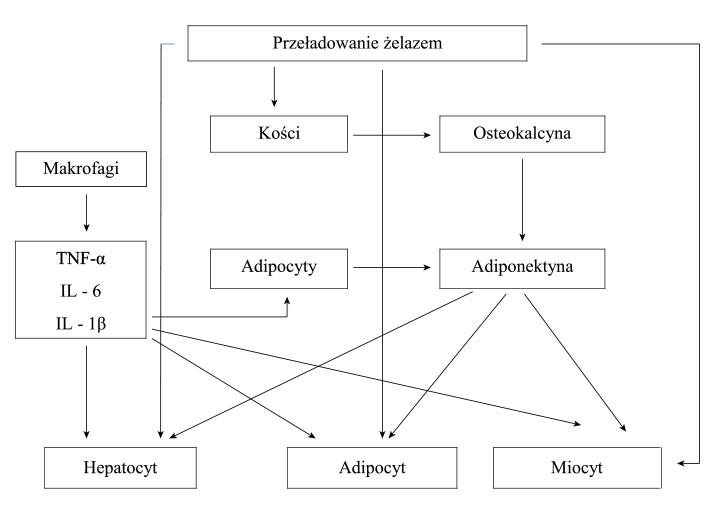
Rycina. Wpływ przeładowania żelazem na narastanie insulinooporności (według 16 w modyfikacji własnej).
Podsumowując zatem, przeładowaniu żelazem towarzyszy z jednej strony upośledzenie czynności komórek beta z powodu niskiej aktywności enzymów antyoksydacyjnych w tych komórkach oraz hamowanej przez podwyższony poziom żelaza HIF-1?, z drugiej zaś nasilenie insulinooporności z powodu wpływu podwyższonego poziomu żelaza na: zwiększenie aktywności prozapalnej makrofagów, zmniejszenie syntezy osteokalcyny przez komórki kostne oraz zahamowanie fosforylacji receptorów insulinowych w tkankach obwodowych. Oba te zaburzenia powodują zwiększone ryzyko rozwoju nietolerancji glukozy i cukrzycy. Nabiera to szczególnego znaczenia w sytuacji, kiedy u chorego stwierdza się już inne zaburzenia mogące w istotny sposób wpływać na czynność komórek beta trzustki czy też na insulinowrażliwość.
Podsumowanie
Na świecie z powodu cukrzycy cierpi coraz więcej osób. Największą część pacjentów stanowią chorzy z typem 2 cukrzycy. W jej patogenezie istotną rolę odgrywają zaburzenia czynności komórek beta oraz insulinooporność.
Do prawidłowego funkcjonowania komórek beta niezbędna jest prawidłowa gospodarka żelazowa. W warunkach fizjologicznych żelazo bierze udział w transporcie elektronów w mitochondriach komórek beta, które są niezbędne dla syntezy ATP i wtórnie do pobudzenia przez glukozę syntezy insuliny. Żelazo bierze także udział w procesie syntezy wolnych rodników tlenowych, które są niezbędne dla inicjacji syntezy insuliny. Przeładowanie żelazem powoduje pogorszenie czynności tychże komórek oraz nasila insulinooporność, co z kolei powoduje zaburzenia gospodarki węglowodanowej, a w konsekwencji prowadzi do rozwoju stanów przedcukrzycowych oraz cukrzycy.
Piśmiennictwo
Simox J.A., McClain DA.: Iron and diabetes risk, Cell Metab, 2013, 17(3), 329341.
Trousseau A.,: Glycosurie, diabete sucre. Clinique Medical de I?Hotel-Dieu de Paris, 1865, 2, 663-698.
Opie E.L.: A case of haemochromatosis the relation of haemochromatosis to bronzed Diabetes, J Exp Med, 1899, 4, (3-4), 279-306.
O?Sullivan E.P., McDermott J.H., Murphy M.S., Sen S., Walsh C.H.: Declining prevalence of diabetes mellitus in hereditary haemochromatosis ? the result of earlier diagnosis, Diabetes Res Clin Pract, 2008, 81 (3)316-320.
Dymock I.W., Cassar J., Pyke D.A., Oakley W.G., Williams R.: Observations on the pathogenesis, complications and treatment of diabetes in 115 causes of haemochromatosis, Am J Med, 1972, 52 (2) 203-210.
Borgna-Pignatti C., Rugolotto S., De Stefano P. i wsp.: Survival and complicationsin patients with thalassemia major treated with transfusion and deferoxamine, Hematologica, 2004, 89 (10), 1187-1193.
Marsella M., Borgna-Pignatti C.: Transfusional iron overload and iron chelation therapy in thalassemia major and sickle cell disease, Hematol Oncol Clin N Am, 2014, 28 (4), 703-727.
Orban E., Schwab S., Thorand B., Huth C.: Association of iron indices and type 2 diabetes: a meta-analysis of obsewrvation studies, Diabetes Metab Res Rev, 2014, 30 (5), 372-394.
Lemandez-Real J.M., Mercader J.M., Ortega F.J. i wsp.:Transferrin receptor ? 1 gene polymorphisms are associated with type 2 diabetes, Eur J Clin Invest, 2010, 40 (7), 600-607.
Qi L., Meigs J., Manson J.E. i wsp.: HFE genetic variability, body iron stores, and the risk of type 2 diabetes in U. S. women, Diabetes, 2005, 54 (12), 3567-3572.
Bao W., Song F., Li X i wsp.: Association betweenheme oxygenase-1 gene promotor polymorphisms nad type 2 diabetes mellitus: a HuGE review and meta ? analysis, Am J Epidemiol, 2010, 172 (6), 631-636.
Lenzen S., Drinkgem J., Tiedge M.: Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tisasues, Free Radic Biol MOl, 1996, 20 (3), 463-466.
Cheng K., Ho K., Stokes R. i wsp.: Hypoxia-inducible factor-1alpha regulates betacell function in mouse and human islets, J CLin Invest, 2010, 120 (6)2171-2183.
Juanola-Falgarona M., Candido-Fernandez J., Salas-Salvado J. i wsp.: Association between serum ferritin and osteocalcin as a potential mechanism explaining the iron-induced insulin resistance, Plos One, 2013, 8 (10), e76433.
Lee N.K., Sowa H., Hinoi E. i wsp.: Endocrine regulation of Energy metabolism by the skeleton, Cell 2007, 130 (3), 456-469.
Wang X., Fang X., Wang F.: Pleiotropic actions of iron balance in diabetes mellitus, Res Endocr Metab Disord, 2015, 16, 15-23.